Poster Presentation RACI Biomolecular Division Conference 2013
Molecular Modeling of Calpain Inhibitors (#80)
Cataract is the leading cause of preventable blindness worldwide according to the World Health Organization. It is a progressive disease that forms cloudiness in the lens of the eye to impair vision and is a major health concern. In recent research, a link has been found between the formation of cataract and the over-activation of the calpain II enzyme. Calpain II is a cysteine protease that requires calcium ions for activation and it is involved in a wide variety of physiological roles due to its ubiquitous nature1. An in vitro model has shown that inhibition of calpain II in lenses has a significant impact on reducing opacification2.
New calpain inhibitors synthesised by Prof Andrew Abell’s group have a unique feature that utilises a large macrocyclic ring with a dipeptide backbone3. Additionally, an aldehyde warhead is present that forms a covalent bond with cysteine in the active site of calpain. This project aims to analyse the new macrocyclic inhibitors by using various computational methods such as docking studies, pharmacophore modelling, ligand-based screening and structure activity relationships.
The binding mode for calpain inhibitors have been mapped as shown in Figure 1. The hydrogen bonds are important for binding as it this enables the covalent binding at Cys105 by aligning the compound in a β-strand formation. Comparisons of the endogenous calpain inhibitor calpastatin with the new macrocyclic variants show similar binding modes. Docking studies also has shown that there is a possible π-π stacking interaction at Trp214 with the macrocyclic inhibitors in addition to the pre-existing binding features. This new potential binding site, termed S4, can be utilised to improve the potency and selectivity of calpain inhibitors.
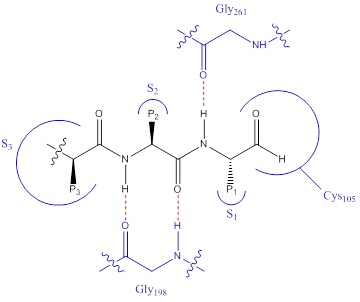
Figure 1. Binding mode for dipeptide calpain inhibitors. Gly198 and Gly261 form hydrogen bonds with the amides in a β-strand conformation.
- Selvakumar, P. and R.K. Sharma, Involvement of calpain in colorectal adenocarcinomas (review). Experimental and Therapeutic Medicine, 2010. 1(3): p. 413-417.
- Abell, A.D., et al., Investigation into the P3 binding domain of m-calpain using photoswitchable diazo- and triazene-dipeptide aldehydes: New anticataract agents. Journal of Medicinal Chemistry, 2007. 50(12): p. 2916-2920.
- Pehere, A.D., et al., Synthesis and Extended Activity of Triazole-Containing Macrocyclic Protease Inhibitors. Chemistry – A European Journal, 2013: p. n/a-n/a.