Poster Presentation RACI Biomolecular Division Conference 2013
Insights into the importance of DFD-motif and insertion I1 in stabilizing the DFD-out conformation of Mnk2 kinase (#96)
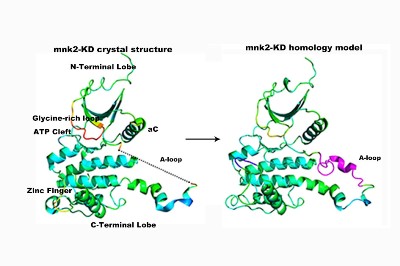
Recent studies have suggested that human mitogen-activated protein kinases (MAPK)-interacting kinases (Mnk)-mediated eukaryotic initiation factor 4E (eIF4E) phosphorylation is crucial for oncogenic activity.1,2 However, Mnk is found to be redundant in the development of normal cells.3 Therefore, targeting Mnk may provide a therapeutic platform for non-toxic anticancer drug development. Mnk proteins possess two distinct features: (1) a unique DFD (Asp-Phe-Asp) motif that replaces the DFG motif found typically in other protein kinases and (2) specific short sequences (the so-called ‘insertion I1’) found in the activation loop.4 Generally, protein kinases adopt the active conformation (i.e. DFG-in) where ATP binding is compatible. Crystallographic studies of Mnk1/2 have revealed that this protein preferentially adopts the inactive conformation (i.e. DFD-out), in which residue Phe228 flips into the ATP binding pocket in the absence of ligands.5 This characteristic is rarely observed in the protein kinome and thus has attracted considerable interest in the design of highly selective Mnk inhibitors. It is believed that the interconnection between the inactive and active conformational change is controlled by the DFG motif in many kinases6 but how a single change in residue (glycine to aspartic acid) impacts on the protein conformation has been left unanswered. Does the specific ‘insertion I1” also play a role in stabilizing the inactive conformation? This work presents for the first time the applicability of 3D models of Mnk2 protein in studying conformational change by utilising high temperature molecular dynamics simulations. Results support previous findings that the native Mnk2 kinase is in favour of an inactive DFD-out conformation. Residue Lys234 of insertion I1 plays a key role in stabilizing the DFD-out conformation through salt bridge formation with Asp226. Understanding the mechanism behind these dynamic processes will aid in the rational design of more selective Mnk2 inhibitors.
- Li, Y.; Yue, P.; Deng, X.; Ueda, T.; Fukunaga, R.; Khuri, F. R.; Sun, S. Y. Protein phosphatase 2A negatively regulates eukaryotic initiation factor 4E phosphorylation and eIF4F assembly through direct dephosphorylation of Mnk and eIF4E. Neoplasia 2010, 12, 848-855.
- Waskiewicz, A. J.; Flynn, A.; Proud, C. G.; Cooper, J. A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J 1997, 16, 1909-1920.
- Konicek, B. W.; Stephens, J. R.; McNulty, A. M.; Robichaud, N.; Peery, R. B.; Dumstorf, C. A.; Dowless, M. S.; Iversen, P. W.; Parsons, S.; Ellis, K. E.; McCann, D. J.; Pelletier, J.; Furic, L.; Yingling, J. M.; Stancato, L. F.; Sonenberg, N.; Graff, J. R. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res 2011, 71, 1849-1857.
- Jauch, R.; Jakel, S.; Netter, C.; Schreiter, K.; Aicher, B.; Jackle, H.; Wahl, M. C. Crystal structures of the Mnk2 kinase domain reveal an inhibitory conformation and a zinc binding site. Structure 2005, 13, 1559-1568.
- Jauch, R.; Cho, M. K.; Jakel, S.; Netter, C.; Schreiter, K.; Aicher, B.; Zweckstetter, M.; Jackle, H.; Wahl, M. C. Mitogen-activated protein kinases interacting kinases are autoinhibited by a reprogrammed activation segment. EMBO J 2006, 25, 4020-4032.
- Shan, Y.; Seeliger, M. A.; Eastwood, M. P.; Frank, F.; Xu, H.; Jensen, M. O.; Dror, R. O.; Kuriyan, J.; Shaw, D. E. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc Natl Acad Sci U S A 2009, 106, 139-144