Oral Presentation RACI Biomolecular Division Conference 2013
Trapped in the act: The α4-α4 interface of α4β2 nicotinic acetylcholine receptors is an additional binding site for methyllycaconitine (#15)
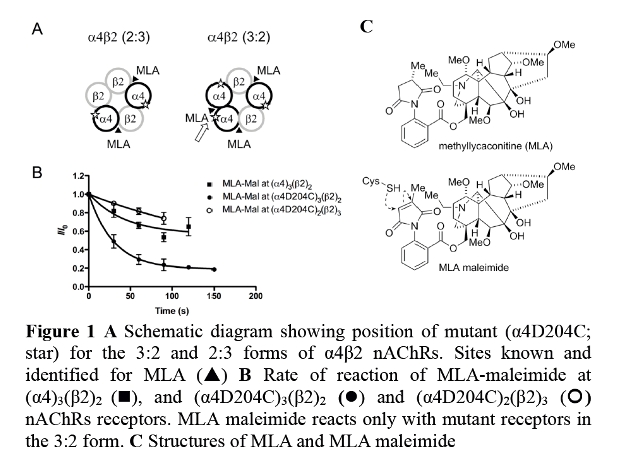
Nicotinic acetylcholine receptors (nAChRs) are ion channels that are widely expressed in the brain, and implicated in nicotine addiction, and memory and learning. The α4β2 nAChR subtype exist in two stoichiometries, (α4)2(β2)3 and (α4)3(β2)2, that differ in sensitivity to the natural agonist, acetylcholine (ACh) and in the current that passes through each channel. Little is known about the effect of stoichiometry on ligand function, or where ligands bind. Recently ACh was found to bind at an additional binding site, located at the α4-α4 interface.1 This site is relatively unexplored and agents that selectively target this site will differentiate between receptor stoichiometry. Methyllycaconitine (MLA) is believed to be a competitive antagonist of nAChRs. Using two-electrode voltage clamp techniques, we evaluated MLA at rat (α4)2(β2)3 and (α4)3(β2)2 nAChRs expressed in Xenopus oocytes. We demonstrate that the inhibition of MLA is either competitive or noncompetitive depending on the stoichiometry tested. Using homology modelling, we identified an amino acid residue, aspartate at position 204 (D204) located on the α4 subunit that can interact with the succinimide group of MLA. After synthesizing a thiol-reactive analogue, MLA-maleimide,2 and mutating the α4 subunit residue to a cysteine (α4D204C), we measured the rate of trapping of MLA-maleimide. The rate of reaction is measured as a reduction in ACh-elicited currents. It was found that the thiol-reactive probe, MLA-maleimide was trapped only in receptors composed of the (α4)3(β2)2 stoichiometry and not the (α4)2(β2)3 stoichiometry, indicating that MLA binds to the α4-α4 interface of the (α4)3(β2)2. Consistent with other studies, we propose that the α4-α4 interface is a structural target for potential therapeutics that modulate (α4)3(β2)2 nAChRs.
- Harpsøe, K.; Ahring, P. K.; Christensen, J. K.; Jensen, M. L.; Peters, D.; Balle, T. J. Neurosci. 2011, 31, 10759
- Ambrus, J. I.; Halliday, J. I.; Kanizaj, N.; Absalom, N.; Harpsøe, K.; Balle, T.; Chebib, M.; McLeod, M. D. Chem Commun (Camb). 2012, 48, 6699.